Managing complex workflows and dependencies in scientific computing can be challenging. Conda helps by managing dependencies and environments, while Apptainer (formerly Singularity) allows for containerized execution, especially in HPC environments. Snakemake, a powerful workflow management system, can integrate these tools to ensure reproducibility and scalability. Additionally, using Apptainer can significantly improve small file performance on software-defined storage systems like CEPHFS. This guide will walk you through converting a Conda environment into an Apptainer image using Snakemake.
Prerequisites
Conda: Make sure Conda is installed.
Apptainer or Singularity: Install Apptainer by following the official guide.
Snakemake: Ensure Snakemake is installed.
Step-by-Step Guide
1. Generate a Dockerfile with Snakemake
First, use Snakemake to generate a Dockerfile for your workflow:
snakemake --containerize > Dockerfile
This command creates a Dockerfile that includes all the necessary dependencies for your Snakemake workflow. Please make sure your workflow itself does not print any text.
update 2025-11-05: Ensure to use snakemake 9.13.3 or greater to include all enviroments
4. Build a Docker container and convert it to Apptainer.
Build in the same dir as the dockerfile the container:
In the past it was possible to convert a Dockerfile to an Apptainer recipe file with the help of Spython. The benefit was that no sudo rights were needed. However, this does not work at the moment.
5. Add container to Snakefile
Tell SnakeMake where to find the container. Add the following to your snakemake file in a global part of the workflow:
containerized: "/path/to/container.sif "
Please note, do use containerized, not conainerize.
6. Run Your Snakemake Workflow with Apptainer
Finally, run your Snakemake workflow using the generated Apptainer image. Ensure the necessary directories are bound to the Apptainer container using the APPTAINER_BIND environment variable:
This command runs the Snakemake workflow, producing the desired VCF file while leveraging the Apptainer container for execution. For snakemake >8 use. snakemake -c1 --software-deployment-method conda apptainer
Addressing Small File Performance Issues
Using Apptainer containers can help alleviate performance issues associated with small files on software-defined storage systems like CEPHFS. These systems often struggle with the overhead of managing numerous small files, leading to degraded performance. By containerizing your environment, you encapsulate your dependencies and binaries into a single image file. This reduces the number of small files accessed directly from the storage system, thus improving I/O performance and overall workflow efficiency.
Conclusion
By following these steps, you can seamlessly integrate Conda environments into Apptainer images using Snakemake, which not only ensures reproducibility and scalability but also addresses small file performance issues on software-defined storage systems like CEPHFS. This workflow enhances your computational environment's portability and efficiency, especially in HPC settings. If you encounter any issues or have questions, feel free to reach out for assistance. Happy computing!
diff will give one line of output that is different: the name and version of the aligner program is different. As expected!
But not everything is still the same! Let's have a look at what kind of hardware is needed to run bwa-mem2.
An Intel or AMD proccesor
Memory:
54 GB or more. This is about 9 times more than bwa.
add memory to this for tools that also pipe data in and out of bwa. For instance picard/samtools sort, samtools fastq etc
Disk space:
index/reference files are 89GB. This is 20 times larger than for bwa.
can be trimmed down with 36-42GB.
How does indexing work with bwa-mem2?
Indexing with bwa and bwa-mem2 both need to index a reference. Indexing takes a while.
Example:
bwa-mem2 index hs38DH_phix.fasta
This is a single-threaded program and takes more than 64GB memory. An error is given when there is not enough memory.
The output is large. An input fasta file of 3.1GB generates for BWA 4.5GB of index and reference files, while bwa-mem2 generates a 89GB files. The numbers(1️⃣ 2️ ⃣) after the listing correspond with which version of bwa uses a file.
When you call bwa-mem2, the program selects automatically the optimized binary for the instruction set. Practically all CPU have a fitting binary
Instruction set
Intel first architecture (year)
AMD first architecture (year)
SSE4.1
Penryn (2008)
Bulldozer (2011)
AVX2
Haswell (2013)
Excavator (2015)
AVX512-BW
Skylake-SP, Skylake-X (2017)
NA
Overview of optimised binaries with the first hardware architecture supporting the instruction set for Intel and AMD CPU/
Note: instructions set works better/faster in newer architecture e.g. AMD ZEN2 can process 2 times more AVX2 instructions the ZEN 1, Haswell downclocks CPU when using AVX2
Is there a difference in memory usage between bwa and bwa-mem2?
bwa-mem2 needs 9 times more memory: 51,5GB while bwa needs 5.7GB. While bwa runs fine on a solid laptop, bwa-mem2 will not run on an average laptop or desktop. Aditional threads consume an additional ~15MB, which is not an issue when using more threads.
In practice, you will need also some additional memory for programs handling input and output of bwa. For example, as input samtools fastq, or as output picard/sambamba/samtools sort or samtools view.
How does bwa-mem2 output look like?
The output (header) prints CPU mode used and other info:
$bwa-mem2-2.0_x64-linux/bwa-mem2 mem -K 10000000 -t 8 hs38DHphix/hs38DH_phix.fasta 1_1.25m.fq 2_1.25m.fq > /dev/null
-----------------------------
Executing in AVX2 mode!!
-----------------------------
* Ref file: hs38DHphix/hs38DH_phix.fasta
* Entering FMI_search
* Index file found. Loading index from hs38DHphix/hs38DH_phix.fasta.bwt.8bit.32
* Reference seq len for bi-index = 6434704607
* Count:
0, 1
1, 1882207599
2, 3217352304
3, 4552497009
4, 6434704607
* Reading other elements of the index from files hs38DHphix/hs38DH_phix.fasta
* Index prefix: hs38DHphix/hs38DH_phix.fasta
* Read 3171 ALT contigs
* Done reading Index!!
* Reading reference genome..
* Binary seq file = hs38DHphix/hs38DH_phix.fasta.0123
* Reference genome size: 6434704606 bp
* Done reading reference genome !!
------------------------------------------
1. Memory pre-allocation for Chaining: 139.3584 MB
2. Memory pre-allocation for BSW: 1916.9362 MB
3. Memory pre-allocation for BWT: 618.5134 MB
------------------------------------------
* Threads used (compute): 8
* No. of pipeline threads:
bwa-mem2 output body
bwa-mem2 loves to chat during work (just like bwa mem):
[0000] read_chunk: 10000000, work_chunk_size: 10000200, nseq: 66668
[0000][ M::kt_pipeline] read 66668 sequences (10000200 bp)...
[0000] Reallocating initial memory allocations!!
[0000] Calling mem_process_seqs.., task: 0
[0000] 1. Calling kt_for - worker_bwt
[0000] read_chunk: 10000000, work_chunk_size: 10000200, nseq: 66668
[0000][ M::kt_pipeline] read 66668 sequences (10000200 bp)...
[0000] 2. Calling kt_for - worker_aln
[0000] Inferring insert size distribution of PE reads from data, l_pac: 3217352303, n: 66668
[0000][PE] # candidate unique pairs for (FF, FR, RF, RR): (10, 27421, 9, 2)
[0000][PE] analyzing insert size distribution for orientation FF...
[0000][PE] (25, 50, 75) percentile: (269, 328, 391)
[0000][PE] low and high boundaries for computing mean and std.dev: (25, 635)
[0000][PE] mean and std.dev: (304.67, 102.65)
[0000][PE] low and high boundaries for proper pairs: (1, 757)
[0000][PE] analyzing insert size distribution for orientation FR...
[0000][PE] (25, 50, 75) percentile: (363, 417, 486)
[0000][PE] low and high boundaries for computing mean and std.dev: (117, 732)
[0000][PE] mean and std.dev: (424.09, 96.84)
[0000][PE] low and high boundaries for proper pairs: (1, 855)
[0000][PE] skip orientation RF as there are not enough pairs
[0000][PE] skip orientation RR as there are not enough pairs
[0000][PE] skip orientation FF
[0000] 3. Calling kt_for - worker_sam
etc...
bwa-mem2 output: tail
Gives timing and setting. The reading of the index files takes about 50 seconds of 46GB of files(or 860MB/s). Keeping index files on slow storage will slowdown the aligning to start
[0000] Computation ends..
No. of OMP threads: 8
Processor is runnig @3600.224460 MHz
Runtime profile:
Time taken for main_mem function: 98.06 sec
IO times (sec) :
Reading IO time (reads) avg: 0.98, (0.98, 0.98)
Writing IO time (SAM) avg: 0.50, (0.50, 0.50)
Reading IO time (Reference Genome) avg: 6.39, (6.39, 6.39)
Index read time avg: 43.00, (43.00, 43.00)
Overall time (sec) (Excluding Index reading time):
PROCESS() (Total compute time + (read + SAM) IO time) : 47.05
MEM_PROCESS_SEQ() (Total compute time (Kernel + SAM)), avg: 46.97, (46.97, 46.97)
SAM Processing time (sec):
--WORKER_SAM avg: 14.55, (14.55, 14.55)
Kernels' compute time (sec):
Total kernel (smem+sal+bsw) time avg: 32.31, (32.31, 32.31)
SMEM compute avg: 17.48, (17.63, 17.32)
SAL compute avg: 1.96, (1.98, 1.93)
BSW time, avg: 9.81, (9.89, 9.76)
Total re-allocs: 4433373 out of total requests: 16786079, Rate: 0.26
Important parameter settings:
BATCH_SIZE: 512
MAX_SEQ_LEN_REF: 256
MAX_SEQ_LEN_QER: 128
MAX_SEQ_LEN8: 128
SEEDS_PER_READ: 500
SIMD_WIDTH8 X: 32
SIMD_WIDTH16 X: 16
AVG_SEEDS_PER_READ: 64
Does bwa-mem2 require all reference files?
Reference files are big for bwa-mem2, but luckily it does not need all files: The SSE binary uses *.bwt.2bit.64 AVX and the AVX512 and AVX2 uses the *.bwt.8bit.32. Removing the unneeded file saves 36-42GB. BWA-mem2 and BWA do not need the FASTA file to perform aligning, but downstream tools often do.
Keep in mind you still need 52 GB of reference data: this can be a problem with provision tools like CVMFS since the local cache might be too small.
bwa-mem2 is developed by Intel, but does it work on an AMD CPU?
Yes, but it can not run the AVX512-bw binary since AMD does not have this instruction set. However, if you compare AVX2 binary running on AMD the results look good.
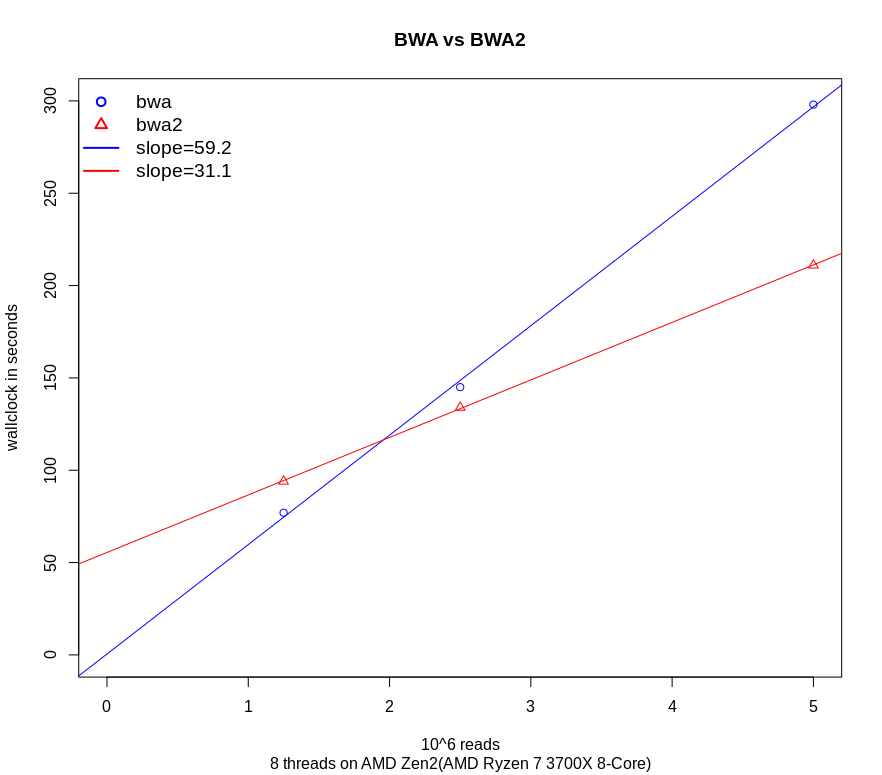
For a benchmark, I used an AMD Zen 2 processor (3700x, 8 cores, 64GB memory) with paired-end reads of 150 base pairs long: I created 3 datasets with a different amount of reads (total amount of reads 1.250.00, 2.500.00 and 5.000.00 ). I ran bwa and bwa-mem2 with 8 threads and plotted them. At first, I was a bit disappointed: with 2 smallest dataset/ bwa-mem2 was slower or a little bit faster. This was caused by the time to read the index/reference files. The time to read these files is reported in the tail of the output of BWA-mem2.
When fitting a linear regression to the timings the time of loading is the same as the intercept (about 50 seconds). In real life most human datasets are bigger: a 30x coverage file needs 124 more reads than this example file and the loading time is not an issue anymore. So comparing the slopes/beta makes more sense than comparing the real timings with a relatively small amount of reads. Where bwa has a slope of 59.2seconds/ million reads bwa-mem2 had a slope of 31.1seconds/million reads. This is a speedup of 1.90. This speedup between AVX2 on Intel is comparable with AVX2 on AMD when comparing the results at the bottom of https://github.com/bwa-mem2/bwa-mem2/.
How well is bwa-mem2 parallelized ?
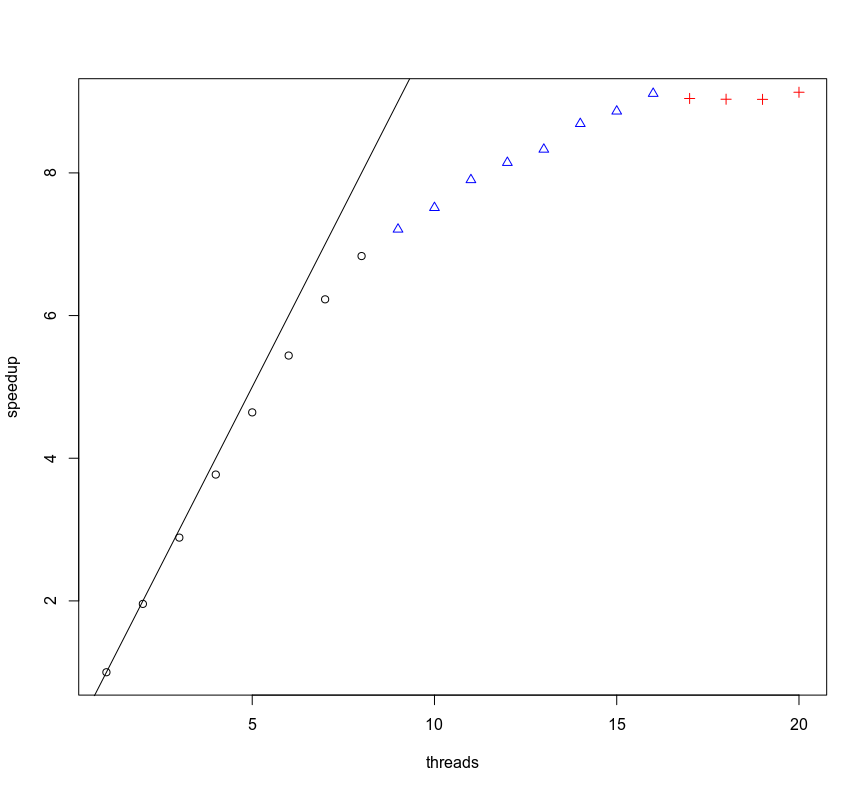
In a parameter sweep on the number of threads (see plot) the speedup follows the speed up nice the amount threads until the "real" number of cores run out: with 8 cores the speedup is 6.83. When adding more thread SMT(hyperthreading is Intel's proprietary implementation) handles this well: when running with 16 threads the speedup is 9.11. Adding more threads than the SMT cores does not improve the speed.
Ran BWA2-mem-2 on 1.250.00 pairs with 1 to 20 threads on AMD Zen 2 processor (3700x, 8 cores, 64GB memory). To prevent benchmarking the CPU the frequency boost was disables with "echo 0 >/sys/devices/system/cpu/cpufreq/boost" Black circles threads use real cores, blue triangles use SMT cores and red cross are oversubscribed. Blackline is optimal speedup of ratio 1.00. Loading the reference and index are subtracted from walltime.
Do index/reference files compress easily?
When working with an object store or central storage with a relatively slow connection to the worker node and a fast local/ scratch storage it might be worth compressing the files.
I compressed the two biggest files with Zstd with level 3.:
On the reference file (*.0123) we save about 72% or 4.6 GB. Extracting this file to /dev/null took about 7 seconds. If the speed of the central storage is slower than 650 megabyte per second, it is advisable to compress when there is a fast scratch storage. Best practice is to uncompressed directly and not copy the file first, because this will slow down the scratch storage due to simultaneous read and write activity.
For the larger *.bwt.2bit.64 compression ratio is much lower : only 24,5% or 9.5 GB is saved. However, Extracting takes about only 23 seconds. If speeds of central storage is slower the 9.5GB/23s~400 it is worth compressing.